Duplicate read pairs can arise in sequencing data via several mechanisms.
Biological duplicates are sequences that arise from different source nucleic-acid molecules that genuinely have the same sequence; these tend to arise when a particular gene or taxon is both extremely abundant and has low sequence diversity. In our case, the most likely cause of biological duplicates are ribosomal RNAs.
Technical duplicates, meanwhile, arise when the same input molecule produces multiple reads. Subgroups of technical duplicates include PCR duplicates arising from amplification of a single input sequence into many library molecules, and several forms of sequencing duplicates arising from errors in the sequencing process. For example, Illumina sequencing on unpatterned flow cells can give rise to optical duplicates, where a single cluster on the flow cell is falsely identified as two by the base-calling algorithm.
In general, we want to remove or collapse technical duplicates, while retaining biological duplicates. Unfortunately, in the absence of UMIs, there’s generally no way to distinguish biological and PCR duplicates; however, many forms of sequencing duplicates can often be identified from the sequence metadata provided in the FASTQ file.
A number of tools are available that attempt to remove some or all of the duplicate sequences in a file. Some of these use cluster positioning information to distinguish sequencing duplicates from other duplicates, while others identify duplicates based purely on their base sequence. In the latter case, the tricky part is identifying the correct threshold for duplicate identification. Due to sequencing errors, requiring perfect base identity between two reads in order to designate them as duplicates often results in true duplicates surviving the deduplication process. On the other hand, designating two reads as duplicates based on too low a sequence identity (or too short a subsequence) will result in spurious deduplications that needlessly throw away useful data.
At the time of writing, our standard sequencing pipeline carries out deduplication very late in the process, during generation of JSON files for the dashboard (i.e. after generating clade counts). Read pairs are identified as duplicates if they are identical in the first 25 bases of both the forward and the reverse read, requiring 50nt of matches overall.
I wanted to see if there was a widely-used read deduplication tool that we could apply to our pipeline, ideally early on as part of sample preprocessing. I started out comparing four approaches, before fairly quickly cutting down to two:
fastp is a FASTQ pre-processing tool previously investigated here. If run with the --dedup`flag, it will remove read pairs that are exact duplicates of one another. As far as I know, fastp doesn’t have the ability to identify or remove inexact duplicates, or to distinguish sequencing duplicates from other duplicates. It thus represents a fast but relatively unsophisticated option.
Clumpify is a tool that was originally developed to reduce space and improve processing speed by rearranging fastq.gz files. It identifies duplicates by looking for reads (or read pairs) that match exactly in sequence, except for a specified number of permitted substitutions. This, to me, is the obvious way to detect duplicates, and this is the only deduplication tool I’ve found that does it this way. It also allows distinguishing of optical vs other duplicates and specific removal of only optical duplicates if desired.
GATK Picard’s MarkDuplicates and samtools markdup are two functions for removing duplicate reads in SAM/BAM files. I tried both, but found them to be slow, confusing, and apparently unable to actually detect any duplicates in the files I ran them on. It’s possible that both of these tools only work on mapped reads (which would make sense given their demand for SAM/BAM files); it’s also possible that I could make them work given more time and effort, but I didn’t want to put this in unless the other tools I found proved inadequate.
To test fastp and Clumpify, I ran them on the same samples I used for looking into ribodetection:
In each case, I tested deduplication at two points in the pipeline: immediately after preprocessing (with FASTP, without deduplication), and following segregation of rRNA reads using bbduk.
1. Johnson (COMO4)
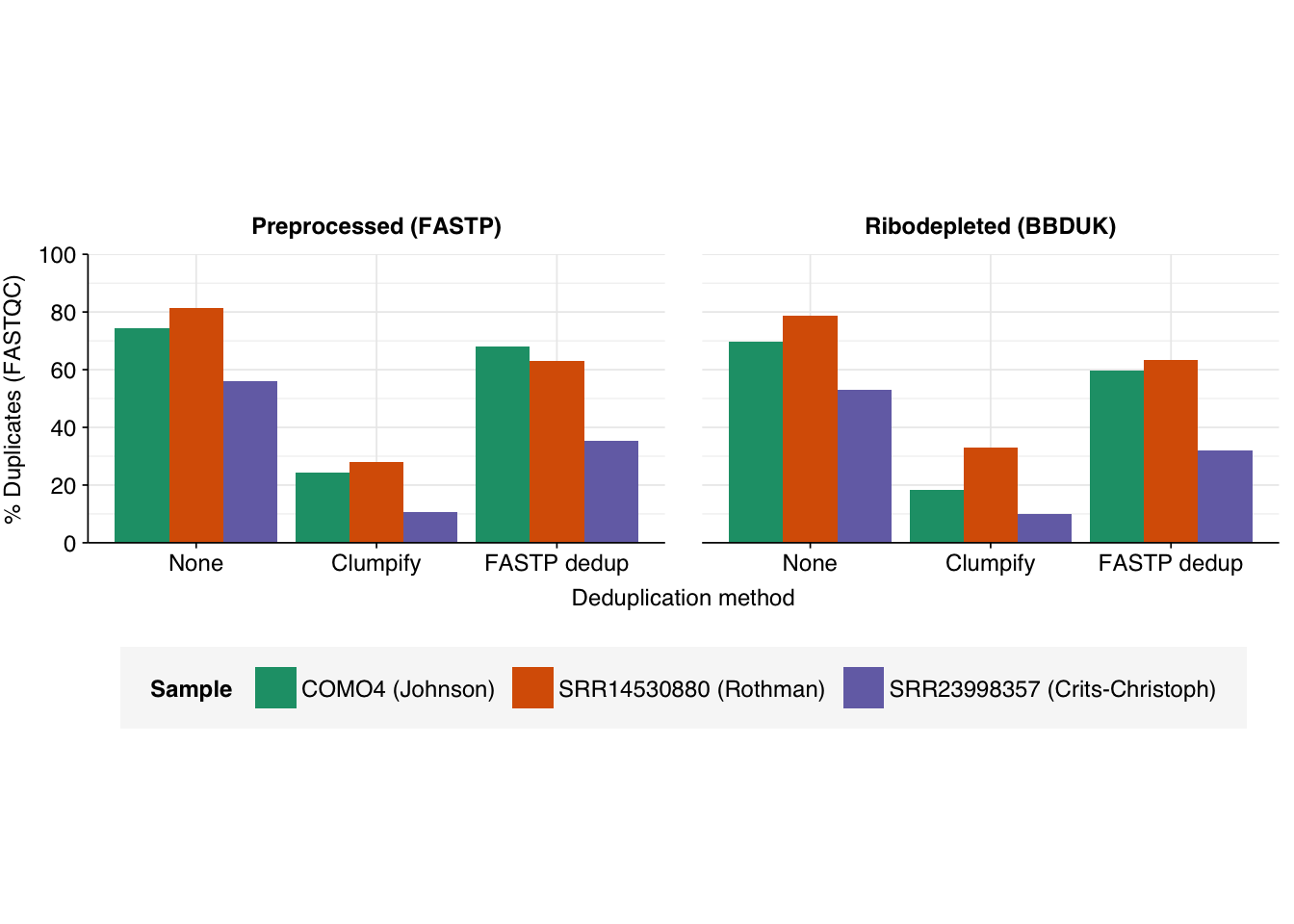
Following fastp preprocessing, the Johnson sample contains 15.25M read pairs, 74.36% of which are identified by FASTQC as duplicates based on the first read in the pair. Among non-ribosomal reads (as identified by bbduk), this number fell to 69.72%, while for ribosomal reads it rose to 79.13%. Needless to say, all of these fail FASTQC’s read-duplication QC test.
Running clumpify on the fastp output took 16 seconds and removed 5.04M read pairs (33.0%) as duplicates, 11576 of which were optical. Running it on the bbduk non-ribosomal output took 9 seconds and removed 2.71M (35.3%) reads as duplicates, 7107 of which were optical. After deduplication with clumpify, FASTQC identifies 24.19% of sequences as duplicates in the post-fastp dataset, and 18.38% in the post-bbduk dataset. In both cases, this is a dramatic reduction, sufficient for FASTQC to now mark the duplication level QC as passing where it was previously failing.
Running fastp with deduplication enabled on the fastp output took 33 seconds and removed 4.21M read pairs (27.6%) as duplicates. Running it on the bbduk non-ribosomal output took 17 seconds and removed 2.33M read pairs (30.4%) as duplicates. In both cases, the number of reads removed is lower than that removed by clumpify, consistent with the fact that fastp requires complete identity between duplicates while clumpify allows a small number of mismatches. For whatever reason, this difference resulted in a dramatic difference in FASTQC quality metrics: after deduplication with fastp, FASTQC identifies 68.09% of reads as duplicates in the full dataset, and 59.78% in the post-bbduk dataset. Both of these are high enough to result in a failure for the corresponding QC test.
Dataset
Dedup Method
FASTQC % Dup
FASTQC Dup Test
Preprocessed (FASTP)
Clumpify
24.19
Passed
Preprocessed (FASTP)
FASTP dedup
68.09
Failed
Ribodepleted (BBDUK)
Clumpify
18.38
Passed
Ribodepleted (BBDUK)
FASTP dedup
59.78
Failed
2. Rothman (SRR14530880)
Following fastp preprocessing, the Rothman sample contains 13.61M read pairs, 81.27% of which are identified by FASTQC as duplicates based on the first read in the pair. Among non-ribosomal reads (6.20M read pairs, as identified by bbduk), this number fell to 78.79%. Both of these failed QC.
Running clumpify on the fastp output took 14 seconds and removed 7.41M read pairs (54.5%) as duplicates. Running it on the bbduk non-ribosomal output took 7 seconds and removed 2.96M read pairs (47.8%) as duplicates. In both cases, trying to specifically detect optical reads caused the program to crash, suggesting that the relevant metadata was unavailable.
Running fastp with deduplication enabled on the fastp output took 26 seconds and removed 6.87M read pairs (50.5%) as duplicates. Running it on the bbduk non-ribosomal output took 14 seconds and removed 2.71M read pairs (43.6%) as duplicates. As before, the number of reads removed was lower than clumpify in both cases.
FASTQ QC results were as follows:
Dataset
Dedup Method
FASTQC % Dup
FASTQC Dup Test
Preprocessed (FASTP)
Clumpify
27.9
Passed
Preprocessed (FASTP)
FASTP dedup
62.9
Failed
Ribodepleted (BBDUK)
Clumpify
33.0
Warning
Ribodepleted (BBDUK)
FASTP dedup
63.2
Failed
As before, the difference is large, with Clumpify performing dramatically better.
3. Crits-Christoph et al. (SRR23998357)
Finally, we have the sample from Crits-Christoph et al. (2021). Following fastp processing, this sample contained 47.47M read pairs, 55.9% of which are identified by FASTQC as duplicates based on the first read in the pair. Among non-ribosomal reads (42.45M read pairs, as identified by bbduk), this number fell to 53.1%. Both of these failed QC.
Running clumpify on the fastp output took 90 seconds and removed 19.25M read pairs (40.6%) as duplicates. Running it on the bbduk non-ribosomal output took 65 seconds and removed 16.49M read pairs (38.8%) as duplicates. As with Rothman, trying to specifically detect optical reads caused the program to crash, suggesting that the relevant metadata was unavailable.
Running fastp with deduplication enabled on the fastp output took 94 seconds and removed 16.58M read pairs (34.9%) as duplicates. Running it on the bbduk non-ribosomal output took 85 seconds and removed 14.12M read pairs (33.3%) as duplicates. As before, the number of reads removed was lower than clumpify in both cases.
FASTQ QC results were as follows:
Both programs perform much better here than for previous samples, with even fastp reducing levels of duplicates low enough to avoid failing. Nevertheless, clumpify continues to perform substantially better.
I was originally planning to do more validation here, but I honestly don’t think it’s required. Of the two methods I managed to successfully apply to these samples, Clumpify is faster; applies an intuitively more appealing deduplication method; removes more sequences; and achieves much better FASTQC results. It’s also very easy to use and configure. I recommend it as our deduplication tool going forward.
In terms of when to apply the tool, I think it probably makes most sense to apply deduplication downstream of counting (if we use Ribodetector) or detecting and filtering (if we use bbduk) ribosomal reads, to avoid spurious filtering of rRNA biological duplicates.
Source Code
---title: "Comparing options for read deduplication"subtitle: "Clumpify vs fastp"author: "Will Bradshaw"date: 2023-10-19format: html: code-fold: true code-tools: true code-link: true df-print: pagededitor: visualtitle-block-banner: black---```{r}#| label: load-packages#| include: falselibrary(tidyverse)library(patchwork)library(ggrepel)source("../scripts/aux_plot-theme.R")```**See also:**- [Comparing FASTP and AdapterRemoval for MGS pre-processing](https://data.securebio.org/wills-public-notebook/notebooks/2023-10-12_fastp-vs-adapterremoval.html)- [Comparing Ribodetector and bbduk for rRNA detection](https://data.securebio.org/wills-public-notebook/notebooks/2023-10-13_rrna-removal.html)Duplicate read pairs can arise in sequencing data via several mechanisms.- **Biological duplicates** are sequences that arise from different source nucleic-acid molecules that genuinely have the same sequence; these tend to arise when a particular gene or taxon is both extremely abundant and has low sequence diversity. In our case, the most likely cause of biological duplicates are ribosomal RNAs.- **Technical duplicates**, meanwhile, arise when the same input molecule produces multiple reads. Subgroups of technical duplicates include **PCR duplicates** arising from amplification of a single input sequence into many library molecules, and several forms of **sequencing duplicates** arising from errors in the sequencing process. For example, Illumina sequencing on unpatterned flow cells can give rise to **optical duplicates**, where a single cluster on the flow cell is falsely identified as two by the base-calling algorithm.In general, we want to remove or collapse technical duplicates, while retaining biological duplicates. Unfortunately, in the absence of UMIs, there's generally no way to distinguish biological and PCR duplicates; however, many forms of sequencing duplicates can often be identified from the sequence metadata provided in the FASTQ file.A number of tools are available that attempt to remove some or all of the duplicate sequences in a file. Some of these use cluster positioning information to distinguish sequencing duplicates from other duplicates, while others identify duplicates based purely on their base sequence. In the latter case, the tricky part is identifying the correct threshold for duplicate identification. Due to sequencing errors, requiring perfect base identity between two reads in order to designate them as duplicates often results in true duplicates surviving the deduplication process. On the other hand, designating two reads as duplicates based on too low a sequence identity (or too short a subsequence) will result in spurious deduplications that needlessly throw away useful data.At the time of writing, our standard sequencing pipeline carries out deduplication very late in the process, during generation of JSON files for the dashboard (i.e. after generating clade counts). Read pairs are identified as duplicates if they are identical in the first 25 bases of both the forward and the reverse read, requiring 50nt of matches overall.I wanted to see if there was a widely-used read deduplication tool that we could apply to our pipeline, ideally early on as part of sample preprocessing. I started out comparing four approaches, before fairly quickly cutting down to two:- fastp is a FASTQ pre-processing tool previously investigated here. If run with the `--dedup`\`flag, it will remove read pairs that are exact duplicates of one another. As far as I know, fastp doesn't have the ability to identify or remove inexact duplicates, or to distinguish sequencing duplicates from other duplicates. It thus represents a fast but relatively unsophisticated option.- [Clumpify](https://www.biostars.org/p/225338/) is a tool that was originally developed to reduce space and improve processing speed by rearranging fastq.gz files. It identifies duplicates by looking for reads (or read pairs) that match exactly in sequence, except for a specified number of permitted substitutions. This, to me, is the obvious way to detect duplicates, and this is the only deduplication tool I've found that does it this way. It also allows distinguishing of optical vs other duplicates and specific removal of only optical duplicates if desired.- GATK Picard's MarkDuplicates and samtools markdup are two functions for removing duplicate reads in SAM/BAM files. I tried both, but found them to be slow, confusing, and apparently unable to actually detect any duplicates in the files I ran them on. It's possible that both of these tools only work on mapped reads (which would make sense given their demand for SAM/BAM files); it's also possible that I could make them work given more time and effort, but I didn't want to put this in unless the other tools I found proved inadequate.To test fastp and Clumpify, I ran them on the same samples I used for looking into ribodetection:| Study | Bioproject | Sample ||-------------------|----------------------------------|-------------------|| Rothman et al. (2021) | [PRJNA729801](https://www.ebi.ac.uk/ena/browser/view/PRJNA729801) | SRR14530880 || Crits-Cristoph et al. (2021) | [PRJNA661613](https://www.ebi.ac.uk/ena/browser/view/PRJNA661613) | SRR23998357 || Johnson (2023) | N/A | COMO4 |In each case, I tested deduplication at two points in the pipeline: immediately after preprocessing (with FASTP, without deduplication), and following segregation of rRNA reads using bbduk.# 1. Johnson (COMO4)Following fastp preprocessing, the Johnson sample contains 15.25M read pairs, 74.36% of which are identified by FASTQC as duplicates based on the first read in the pair. Among non-ribosomal reads (as identified by bbduk), this number fell to 69.72%, while for ribosomal reads it rose to 79.13%. Needless to say, all of these fail FASTQC's read-duplication QC test.Running clumpify on the fastp output took 16 seconds and removed 5.04M read pairs (33.0%) as duplicates, 11576 of which were optical. Running it on the bbduk non-ribosomal output took 9 seconds and removed 2.71M (35.3%) reads as duplicates, 7107 of which were optical. After deduplication with clumpify, FASTQC identifies 24.19% of sequences as duplicates in the post-fastp dataset, and 18.38% in the post-bbduk dataset. In both cases, this is a dramatic reduction, sufficient for FASTQC to now mark the duplication level QC as passing where it was previously failing.Running fastp with deduplication enabled on the fastp output took 33 seconds and removed 4.21M read pairs (27.6%) as duplicates. Running it on the bbduk non-ribosomal output took 17 seconds and removed 2.33M read pairs (30.4%) as duplicates. In both cases, the number of reads removed is lower than that removed by clumpify, consistent with the fact that fastp requires complete identity between duplicates while clumpify allows a small number of mismatches. For whatever reason, this difference resulted in a dramatic difference in FASTQC quality metrics: after deduplication with fastp, FASTQC identifies 68.09% of reads as duplicates in the full dataset, and 59.78% in the post-bbduk dataset. Both of these are high enough to result in a failure for the corresponding QC test.| Dataset | Dedup Method | FASTQC % Dup | FASTQC Dup Test ||----------------------|--------------|--------------|-----------------|| Preprocessed (FASTP) | Clumpify | 24.19 | Passed || Preprocessed (FASTP) | FASTP dedup | 68.09 | Failed || Ribodepleted (BBDUK) | Clumpify | 18.38 | Passed || Ribodepleted (BBDUK) | FASTP dedup | 59.78 | Failed |# 2. Rothman (SRR14530880)Following fastp preprocessing, the Rothman sample contains 13.61M read pairs, 81.27% of which are identified by FASTQC as duplicates based on the first read in the pair. Among non-ribosomal reads (6.20M read pairs, as identified by bbduk), this number fell to 78.79%. Both of these failed QC.Running clumpify on the fastp output took 14 seconds and removed 7.41M read pairs (54.5%) as duplicates. Running it on the bbduk non-ribosomal output took 7 seconds and removed 2.96M read pairs (47.8%) as duplicates. In both cases, trying to specifically detect optical reads caused the program to crash, suggesting that the relevant metadata was unavailable.Running fastp with deduplication enabled on the fastp output took 26 seconds and removed 6.87M read pairs (50.5%) as duplicates. Running it on the bbduk non-ribosomal output took 14 seconds and removed 2.71M read pairs (43.6%) as duplicates. As before, the number of reads removed was lower than clumpify in both cases.FASTQ QC results were as follows:| Dataset | Dedup Method | FASTQC % Dup | FASTQC Dup Test ||----------------------|--------------|--------------|-----------------|| Preprocessed (FASTP) | Clumpify | 27.9 | Passed || Preprocessed (FASTP) | FASTP dedup | 62.9 | Failed || Ribodepleted (BBDUK) | Clumpify | 33.0 | Warning || Ribodepleted (BBDUK) | FASTP dedup | 63.2 | Failed |As before, the difference is large, with Clumpify performing dramatically better.# 3. Crits-Christoph et al. (SRR23998357)Finally, we have the sample from Crits-Christoph et al. (2021). Following fastp processing, this sample contained 47.47M read pairs, 55.9% of which are identified by FASTQC as duplicates based on the first read in the pair. Among non-ribosomal reads (42.45M read pairs, as identified by bbduk), this number fell to 53.1%. Both of these failed QC.Running clumpify on the fastp output took 90 seconds and removed 19.25M read pairs (40.6%) as duplicates. Running it on the bbduk non-ribosomal output took 65 seconds and removed 16.49M read pairs (38.8%) as duplicates. As with Rothman, trying to specifically detect optical reads caused the program to crash, suggesting that the relevant metadata was unavailable.Running fastp with deduplication enabled on the fastp output took 94 seconds and removed 16.58M read pairs (34.9%) as duplicates. Running it on the bbduk non-ribosomal output took 85 seconds and removed 14.12M read pairs (33.3%) as duplicates. As before, the number of reads removed was lower than clumpify in both cases.FASTQ QC results were as follows:| Dataset | Dedup Method | FASTQC % Dup | FASTQC Dup Test ||----------------------|--------------|--------------|-----------------|| Preprocessed (FASTP) | Clumpify | 10.64 | Passed || Preprocessed (FASTP) | FASTP dedup | 35.22 | Warning || Ribodepleted (BBDUK) | Clumpify | 9.89 | Passed || Ribodepleted (BBDUK) | FASTP dedup | 32.07 | Warning |: Both programs perform much better here than for previous samples, with even fastp reducing levels of duplicates low enough to avoid failing. Nevertheless, clumpify continues to perform substantially better.```{r}duplicate_data_path <-"../data/2023-10-19_deduplication/duplicate-data.csv"duplicate_data <-read_csv(duplicate_data_path, show_col_types =FALSE) %>%mutate(sample_display =paste0(sample, " (", dataset, ")"),dedup_method =fct_inorder(dedup_method),sample_display =fct_inorder(sample_display))g_duplicate <-ggplot(duplicate_data, aes(x=dedup_method, y=fastqc_pc_dup, fill=sample_display)) +geom_col(position ="dodge") +scale_y_continuous(name ="% Duplicates (FASTQC)", limits =c(0,100),breaks =seq(0,100,20), expand =c(0,0)) +scale_x_discrete(name ="Deduplication method") +scale_fill_brewer(palette ="Dark2", name ="Sample") +facet_grid(. ~ processing_stage) + theme_baseg_duplicate```# 4. ConclusionI was originally planning to do more validation here, but I honestly don't think it's required. Of the two methods I managed to successfully apply to these samples, Clumpify is faster; applies an intuitively more appealing deduplication method; removes more sequences; and achieves much better FASTQC results. It's also very easy to use and configure. I recommend it as our deduplication tool going forward.In terms of when to apply the tool, I think it probably makes most sense to apply deduplication downstream of counting (if we use Ribodetector) or detecting and filtering (if we use bbduk) ribosomal reads, to avoid spurious filtering of rRNA biological duplicates.